重要提示:
在临床研究中,观察到口服爱谱沙期间,患者可能会出现血小板降低、白细胞减少、中性粒细胞减少、血红蛋白降低等血液学毒性,请在医生指导下使用,并在用药过程中定期进行血常规检查后调整剂量或予以对症治疗。乏力、胃肠道系统的不良反应,如腹泻、食欲下降、恶心、呕吐、消化不良、口腔溃疡也可能在服药期间发生。发热、全身水肿、呼吸道感染、皮疹、瘙痒、过敏性皮炎、低钾、低钙血症、低蛋白血症、头晕、感觉减退、背痛、关节痛、肌痛、心包积液等也在少量患者中观察得到。因此,请在专科医生指导下应用爱谱沙,并在服用爱谱沙治疗期间定期血常规检查。了解以上内容后请阅读下面的内容。





组蛋白去乙酰化酶(HDAC)
染色质的组蛋白乙酰化和去乙酰化是调节基因表达的关键环节之一,而两类酶决定着组蛋白的乙酰化程度,即组蛋白乙酰基转移酶(Histone acetyltransferases,HAT)和组蛋白去乙酰化酶(Histone deacetylases,HDAC)。HAT将乙酰辅酶A的乙酰基部分,转移到染色质核心组蛋白氨基末端上特定赖氨酸(Lys)残基的氨基基团上,从而使氨基上的正电荷被中和,这时DNA分子本身所带有的负电荷有利于DNA构象的展开,使染色质结构变得松弛。这种松弛的结构促进了转录因子及其辅助因子与DNA分子的接触,因此组蛋白的乙酰化可以激活特定基因的转录过程。而HDAC则移去组蛋白Lys残基上的乙酰基,恢复组蛋白的正电性,带正电荷的Lys残基与DNA分子的电性相反,增加了DNA与组蛋白之间的吸引力,染色质结构变得紧密,使转录因子及其辅助因子不易接近转录调控元件,从而抑制基因的转录表达。HDAC还对非组蛋白蛋白质的乙酰化-去乙酰化过程有着重要影响,包括转录因子、信号传导蛋白、DNA修复酶等等,而这些靶蛋白在基因表达的调控方面起着决定性作用。总之,通过对组蛋白及非组蛋白乙酰化过程的影响,HDAC在表观遗传调控方面具有极为重要的作用,而这一调控机制的异常,则与肿瘤的发生和发展则与肿瘤的发生和发展密切相关。
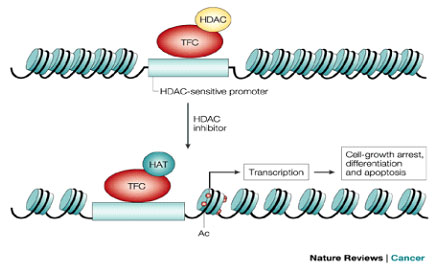
HDAC与基因转录调控
组蛋白去乙酰化酶(HDAC)包括四大类18个不同的亚型(Ⅰ类:HDAC1、2、3、8;II类:HDAC4、5、6、7、9、10;III类:Sirt1-7;IV类:HDAC11),它与组蛋白乙酰基转移酶(HAT)共同调节组蛋白的乙酰化修饰,HAT对组蛋白上特定赖氨酸(Lys)残基进行乙酰化而HDAC负责移除该残基修饰,组蛋白的乙酰化使得染色质结构变得松弛,从而有利于其他DNA结合蛋白(如转录因子等)的结合,因此组蛋白的乙酰化可以激活特定基因的转录过程(染色质的重塑)。同时,HDAC还调节部分非组蛋白类蛋白质底物的乙酰化修饰,如转录调控因子(P53、NF-κB等)、应激反应蛋白(Hsp70/90等)以及细胞结构分子(Tubulin等)等,进一步影响细胞增殖生长及其他生物学过程。
不同HDAC亚型在序列同源性及细胞定位上存在一定差异,其中第I大类HDAC主要在细胞核内行使功能,唯一例外的是HDAC3具有在细胞核与胞浆间移动的能力,其他大类的亚型则主要为细胞核和胞浆穿梭蛋白或定位于胞浆内(如HDAC6、10、11),后者仅对胞浆中的非组蛋白底物发挥乙酰化调节作用。除第I大类HDAC在不同组织细胞中普遍表达外,其他亚型的表达分布具有明显的组织特异性。
不同HDAC亚型调节的生物活性及具体机制目前还不完全清楚,基于已有的研究结果,第I大类的HDAC参与不同的转录抑制复合体形成(HDAC1/2:Sin3、NuRD、CoREST和PRC2等;HDAC3:N-CoR–SMRT等),由此调控不同的靶基因转录及行使不同的下游功能;第II大类中HDAC6和HDAC10主要在胞浆中调节非组蛋白底物的乙酰化,这些蛋白底物中包括多种转录调控因子如β-Catenin、STATs、NF-κB等,它们的乙酰化修饰改变直接影响下游靶基因的转录。应用小分子RNA干扰技术和动物基因敲除技术所得到的研究结果综合显示,I类和II类中的一些HDAC亚型,可能是抗肿瘤作用相关的靶标,因此,HDAC代表了一类具有广泛的生物学功能调节的蛋白家族,参与了体内各种生理病理过程,包括肿瘤。
HDAC与肿瘤免疫
HDAC抑制剂对免疫功能的影响已有大量的研究,由于免疫系统涉及的细胞类型多而复杂,而不同HDAC亚型在其中的调节作用也可能存在明显差异,因此采用不同类型的HDAC抑制剂得出的研究结果往往不一致。因涉及到对不同血系细胞的分化调节、免疫呈递细胞(如Dendritic cells, DCs)以及Treg的活性调节等等,第Ⅰ大类和第Ⅱ大类HDAC抑制剂在亚型选择性上的差异,在对免疫调控方面可能存在相反的作用。
HDAC与肿瘤干细胞及EMT肿瘤的转移和治疗后的复发是临床治疗面临的最大挑战,也是绝大部分(90%)肿瘤患者最终死亡的直接原因。肿瘤的转移涉及到肿瘤细胞从一个病灶迁移到新的环境并复制肿瘤早期生长过程,肿瘤复发同样是从治疗后残留的肿瘤细胞(无论是原位还是异位,具有耐药性特点)开始新的肿瘤形成过程。虽然其中涉及的具体机制目前还不完全清楚,但是两者之间的相似性都提示存在一个共同的机制特点,比较普遍接受的观点是肿瘤干细胞(CSC)在其中的地位。按照现有的定义,肿瘤干细胞是具有独立形成肿瘤的能力、且对药物治疗相对耐受。
在肿瘤转移过程中,肿瘤细胞的表型转换(上皮间充质表型转换,Epithelial-Mesenchymal Transition,EMT)扮演了重要的角色。在肿瘤细胞发生表型转换时,其细胞表面分子的表达发生相应的变化,如上皮钙粘蛋白(E-Cadherin,E-cad)。E-cad是上皮细胞间粘附的主要分子,在肿瘤中,细胞间的粘附作用使肿瘤细胞之间保持密切的接触,一旦肿瘤细胞表面E-cad表达降低或消失,细胞由上皮型变为间充质表型,将导致肿瘤细胞间粘附松散,从而易于从肿瘤组织中脱离继而穿过血管和组织屏障,在其他合适的部位形成新的肿瘤。E-cad在多种不同类型的肿瘤包括肺癌、乳腺癌、前列腺癌、胃癌、结直肠癌、卵巢癌等肿瘤组织中普遍存在低表达或缺失的现象,临床研究发现肿瘤组织中E-cad的表达水平与患者的疗效反应或生存期显著相关性。
虽然相关的研究目前开展较少,但是已有研究显示,HDAC抑制剂可以抑制部分肿瘤干细胞的活性,同时,包括DNA甲基化、组蛋白甲基化、乙酰化等表观遗传调控模式在肿瘤干细胞中的调节活性研究目前也在广泛开展。在最近完成的一项临床研究中,HDAC抑制剂MS-275联合蛋白激酶抑制剂(TKI)厄洛替尼(Tarceva)相比于厄洛替尼单药针对E-cad表达阳性非小细胞肺癌患者取得了显著的生存期改善(OS),HDAC抑制剂对E-cad的表达以及EMT过程的调节作用已经有许多的研究报道,这类药物对E-Cad的表达促进作用可能是上述MS-275联合厄洛替尼临床试验中患者从联合治疗方案中进一步获益的重要原因之一,因此进一步研究E-cad表达在HDAC抑制剂药物临床研究中的疗效相关性具有重要意义。
HDAC与DNA甲基化
组蛋白去乙酰化与DNA甲基化在肿瘤抑制基因的沉默方面具有协同效应。以甲基化胞嘧啶结合蛋白MeCP2为例,当启动子区的CpG发生甲基化时,MeCP2与甲基化的CpG结合后,再与Sin3A结合,进一步与异二聚体Mad/Max形成复合物。该复合物募集HDAC,形成一个共同转录抑制因子。HDAC使组蛋白去掉乙酰基,重塑染色质结构,从而阻碍了基本转录单位蛋白质复合物进入启动子结合位点,导致转录抑制。另外,有实验表明DNA甲基转移酶-1(DNMT-1)能直接与HDAC结合,发挥协同抑制基因转录的作用。
研究表明,组蛋白去乙酰化抑制基因转录的作用依赖于甲基化CpG位点的数目。当发生甲基化改变的启动子区域的CpG位点的数量不足以引起长距离的基因抑制的时候,组蛋白去乙酰化在抑制基因表达上将发挥重要的作用。在这种情况下,用组蛋白去乙酰化酶抑制剂TSA处理可以使因甲基化而静止的基因恢复表达,抑制被解除,说明甲基化所致的染色质结构的紧缩不够稳定,不足以维持这种紧缩的状态。而当发生甲基化的启动子区域的CpG位点的数目增大到可以引起基因广泛静止的阈值的时候,这种抑制的机制将会有明显的不同。此时用TSA处理,转录水平仍明显低于未甲基化的对照,说明在这种情况下,高密度的甲基化使染色质结构高度紧缩并且相当稳定,组蛋白去乙酰化对转录抑制的作用变为次要。
组蛋白去乙酰化酶抑制剂(HDACi)
近年来的研究结果已充分显示,HDAC过度表达或活性异常在白血病和实体肿瘤的发生及发展中起着重要的作用,而抑制HDAC的功能活性则显示出显著的体内外抗肿瘤效果。因此,以HDAC为靶标的抗肿瘤药物研发正在全球范围内充分展开,其中由默克公司开发的Vorinostat(SAHA),在完成II期临床试验后,已于2006年底被美国FDA批准以皮肤T淋巴细胞瘤(CTCL)为适应症而上市应用;HDAC抑制剂Romidepsin(FK228)于2009年被美国FDA批准上市用于CTCL的治疗,于2011年被美国FDA批准上市用于复发性/难治性PTCL的治疗。此外,目前全球范围内还有多个HDAC抑制剂正在临床试验阶段,所涉及的适应症包括了白血病、淋巴瘤和实体瘤(见下表)。
化合物 | 结构类别 | 选择性 | 适应症 | 治疗方式 | 临床阶段 | 适应者 |
| 伏立诺他( Vorinostat SAHA Zolinza) | 羟肟酸类 | I、II类HDAC | CTCL | 单药 | 已上市 | 美国Merck |
| PTCL | 联合用药 | II期 | ||||
弥漫性大B细胞淋巴瘤 | 单药/联合用药 | II期 | ||||
| NSCLC | 联合用药 | III期 | ||||
| 乳腺癌 | 联合用药 | II期 | ||||
| 卵巢癌 | 联合用药 | II期 | ||||
| 前列腺癌 | 联合用药 | II期 | ||||
| 结直肠癌 | 联合用药 | II期 | ||||
| Romidepsin (FK228Istodax) | 环四肽类 | I、II类HDAC | CTCL | 单药 | 已上市 | 美国Celgene |
| PTCL | 单药 | 已上市 | ||||
| 西达本胺 (ChidamideCS055 爱谱沙) | 苯酰胺类 | I类HDAC(对1、3高选择性) | PTCL | 单药 | 2014年12月获CFDA批准 | 中国微芯生物 |
| CTCL | 单药 | 关键II期 | ||||
| NSCLC | 联合用药 | II期 | ||||
| Entinostat (MS-275) | 苯酰胺类 | I类HDAC(对1高选择性) | 霍奇金淋巴瘤 | 单药 | II期 | 德国Bayer和美国Syndax |
| NSCLC | 联合用药 | II期 | ||||
| 乳腺癌 | 联合用药 | II期 | ||||
| 结直肠癌 | 联合用药 | II期 | ||||
| Mocetinostat (MGCD0103) | 苯酰胺类 | HDAC 1, 2, 3, 11 | DLBCL | 单药 | 2014年8月FDA授予孤儿药资格 | 美国Mirati Therapeutics |
| MDS | 单药 | 2014年7月FDA授予孤儿药资格 | ||||
| 膀胱癌 | 联合用药 | II期 | ||||
| Panobinostat (LBH589) | 羟肟酸类 | I、II类HDAC | 多发性骨髓瘤 | 联合用药 | 2015年2月FDA批准 | 瑞士Novartis |
| CTCL | 单药 | II/III期 | ||||
| 霍奇金淋巴瘤 | 单药/联合用药 | II/III期 | ||||
| 乳腺癌 | 联合用药 | II期 | ||||
| Belinostat (PXD101) | 羟肟酸类 | I、II类HDAC | CTCL | 单药 | II期 | 美国uraGen和丹麦TopoTarget |
| CTCL | 单药 | 2014年7月FDA批准 | ||||
| 骨髓增生异常综合征 | 单药 | II期 | ||||
| Resminostat (4SC-201) | 羟肟酸类 | I、II类HDAC | 霍奇金淋巴瘤 | 单药 | II期 | 德国4SC |
| 肝细胞癌 | 单药/联合用药 | II期 |
HDAC抑制剂按化学结构可被分为四大类,即羟肟酸类、环四肽类、短链脂肪酸和苯酰胺类。前三大类化合物抑制I类和II类所有HDAC亚型,属于HDAC非选择性抑制剂,它们之间的差别只是体现在作用强度方面(羟肟酸类和环四肽类的体外有效作用浓度在纳摩尔~微摩尔间,而短链脂肪酸类为毫摩尔浓度)。苯酰胺类HDAC抑制剂则显示出靶标作用的选择性,它们主要抑制I类HDAC(包括HDAC亚型1、2、3,但不抑制HDAC8)和部分IIb类HDAC,对IIa类HDAC无抑制作用。
Vorinostat与Romidepsin在美国的成功上市,标示着HDAC作为新颖药物靶标的概念验证性研究阶段的结束。尽管HDAC抑制剂在血液及淋巴瘤的治疗作用已经得以确认,但针对实体瘤的单药或联合用药结果仍然不理想。随着研究的不断深入,HDAC抑制剂的抗肿瘤作用机制也被逐渐发现,几乎涉及到了肿瘤发生和发展的方方面面,包括诱导肿瘤细胞凋亡、抑制肿瘤细胞周期、诱导肿瘤细胞分化、抑制血管生成、抑制肿瘤转移、调节抗肿瘤免疫功能等,其中一些作用机制已在肿瘤患者上得到确证。

浏览手机网站
浏览微信网站
